About nrseq-fasta-importer¶
The YRC_NRSEQ database¶
The purpose of this application is to parse a FASTA file into the YRC_NRSEQ database. The YRC_NRSEQ database stores non-redundant protein sequences, proteins that reference those sequences (a protein being a given sequence in a particular species), annotations that reference those proteins (names and descriptions from different sources), and annotations for the sources (e.g., NCBI, Uniprot, or individual FASTA files). This is accomplished using four simple tables:
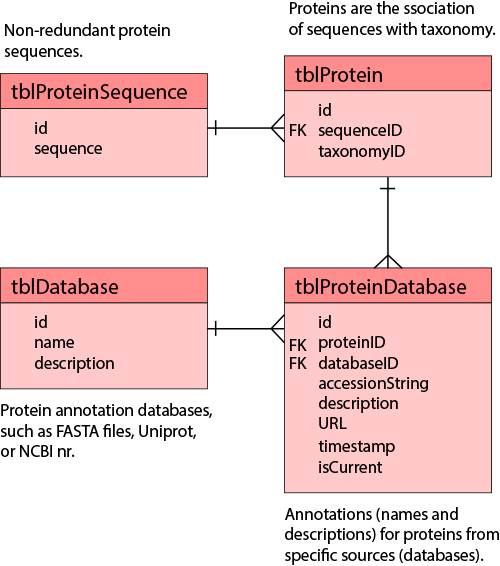
Proteins are regarded simply as a specific protein sequence from a specific organism. This allows for unambiguously determining if two different experiments that may use different naming conventions are referring to the same protein–allowing for the ability to compare and contrast that is independent of the naming convention (e.g., FASTA file) used in the respective experiment. It also allows for knowing the organism associated with a protein in an experiment, and for showing the names or descriptions for those proteins from any parsed naming database.
Why do we have to parse FASTA files?¶
In order to reap the benefits of the YRC_NRSEQ database, a FASTA file must be parsed and inserted into the database. A FASTA file has this basic structure:
>NAME1 Descriptive text
SEQUENCE
>NAME2 Descriptive text
SEQUENCE
>NAME3 Descriptive text
SEQUENCE
In a proteomics experiment, proteins are typically referenced by the names associated with protein sequences in the FASTA file (NAME1, NAME2, and NAME3 in the above example). By parsing the FASTA file in advance, an application built to interface with YRC_NRSEQ knows which protein sequence and organism is being referenced by that name. When combining results from multiple experiments, the application will know if two names are describing the same protein. When showing results, the name from that FASTA file or from a preferred naming database (that has also been parsed into the YRC_NRSEQ) may be shown. And other protein annotations, such as domain predictions, that are associated with YRC_NRSEQ protein IDs may also be shown along with experimental data to provide biological context.
How does parsing work?¶
In general terms, parsing works as follows:
- Validate the FASTA file
- Each FASTA name only appears once
- Each FASTA header has a sequence
- This FASTA file hasn’t already been parsed
- Confirm a NCBI taxonomy ID can be determined for each FASTA header
- If this isn’t possible, import should not proceed
- User input may be required
- This is the trickiest part of the import process and described in more detail below
- For each FASTA entry (header + sequence) in the FASTA file:
Insert an entry for this FASTA file in tblDatabase, get ID
- Get the sequence ID for the current sequence
- If not in the database, insert it and get the resulting ID
Determine the NCBI taxonomy ID
- Determine the protein ID (from sequence ID and taxonomy ID)
- If not in the database, insert it and get the resulting ID
Insert the annotation for this protein from this FASTA file in tblProteinDatabase
How does the taxonomy lookup work?¶
Software, such as the nrseq-fasta-importer web application, may employ any logic to determine the NCBI taxonomy ID that should be associated with a given FASTA header. The logic employed by the nrseq-fasta-importer works roughly as follows:
- Does the FASTA header look like a reversed or shuffled sequence? Use 0 for the taxonomy ID.
- Does the FASTA header contain
Tax_Id=###orTaxonomy_Id=###? Use ###.- Does the FASTA header end with
[species name]? Lookup the taxonomy ID for that species name.- Does the FASTA header contain
organism=species_name? Lookup the taxonomy ID for that species name.- Does the FASTA header contain
OS=speces name? Lookup the taxonomy ID for that species name.- Does the FASTA header name look like a NCBI accession string? If so, lookup the taxonomy ID from NCBI.
- Does the FASTA header name look like a swiss-prot accession string? If so, lookup the taxonomy ID from swiss-prot.
- Does the FASTA header name look like a Uniprot accession string? If so, lookup the taxonomy ID from Uniprot.
- Does the FASTA header name look like a SGD accession string? If so, lookup the taxonomy ID from SGD.
- Does the FASTA header name look like a Wormbase accession string? If so, lookup the taxonomy ID from Wormbase.
- Does the FASTA header name look like a Flybase accession string? If so, lookup the taxonomy ID from Flybase.
What if the taxonomy ID cannot be found?¶
The nrseq-fasta-importer web application provides an interface for providing NCBI taxonomy IDs for any FASTA headers for which it could not be determined. (See Using nrseq-fasta-importer.) If the taxonomy ID cannot be determined for a very large number of entries, several strategies are available:
- Write a script to modify the FASTA file and add
Tax_Id=XXXto each FASTA header, where XXX is the NCBI taxonomy ID. This is often a good choice if your FASTA file is of your own making and the headers require custom taxonomy lookup logic.- Write a new taxonomy lookup module for the nrseq-fasta-importer web application. Although harder, this is often a good choice if the FASTA headers include names or description from a protein naming database you often use or encounter. The authors of nrseq-fasta-importer are happy to work with you to add a new lookup module.
- Write your own parser script. Although not generally recommended, importing into the YRC_NRSEQ database is not conceptually complex and may be the right choice if you are an advanced user and are comfortable with scripting.